2025年12月1日,北京大学未来技术学院、定量生物学中心朱怀球课题组及合作者研究团队在《Nature Communications》发表题为“Deciphering gene redundancy in prokaryotic genomes provides evolutionary insights for pathogenicity and its roles in clinical infections”的最新研究成果。长期以来,人们对真核生物的基因冗余(重复基因)及其重要作用有广泛深入的研究,但对于像病原菌这样基因组紧凑的微生物,基因冗余的规律及作用仍然缺乏系统性的认识。该团队的这一研究通过对22,310个完整原核基因组的大规模分析,从全局视角揭示了基因冗余在微生物中广泛存在的普遍性。尽管原核基因组高度紧凑,冗余基因仍在绝大多数基因组中存在,尤其是病原菌,普遍比其他细菌基因组拥有更高比例的冗余基因。进一步的生态位比较分析表明,冗余基因在海洋、陆地、植物和动物宿主等不同环境中呈现特定的功能倾向,凸显其在微生物环境适应中具有的重要作用。从进化角度来看,冗余基因的数量随系统发育距离增加而扩张,且大多数冗余副本保留完整功能特征:基因的复制常伴随其翻译起始信号的一同复制,假基因化比例极低。这表明冗余并非随机重复,而是微生物维持适应性和功能弹性的关键机制。研究团队对898株Enterobacter cloacae complex(阴沟肠杆菌复合群)的分析显示,通过质粒和水平基因转移频繁获得重金属耐受、毒力因子等冗余基因,病原菌从而强化了在医院环境中的适应与竞争优势。为评估冗余基因在临床感染中的意义,朱怀球教授课题组与合作者浙江大学医学院附属第一医院肖永红教授课题组对ICU(重症监护室)患者来源的69株Acinetobacter baumannii(鲍曼不动杆菌)的全基因组测序进行时间序列分析。结果显示,尿路感染菌株中特异富集的乙醇脱氢酶家族基因frmA与ZADH的双拷贝冗余源自基因岛,并在体内传播过程中长期稳定存在。进一步的动物实验证实,frmA的冗余能显著提升菌株的致病性与生物膜形成能力,揭示了该基因在宿主感染中的直接功能贡献。

长期以来,人们已认识到具有较大基因组的真核生物基因组结构通常更为复杂,由此对真核基因组中冗余基因的产生与功能进行了广泛、深入的系统研究。然而,对于基因组结构更为紧凑、进化压力强烈的原核生物而言,其冗余基因的分布特征、功能作用及进化轨迹却长期缺乏系统性研究。近年来,朱怀球教授团队针对这一问题进行了系统、深入的研究。
1. 基因冗余在原核生物特别是病原菌中普遍存在,且与生态位适应显著相关
研究团队首先构建了高通量分析的流程工具SoDpipe。运用这一工具,对来自RefSeq的22,310个完整原核生物基因组进行系统分析,建立了目前规模最大的原核生物冗余基因图谱。分析显示,97%以上的原核基因组都含有冗余基因,尽管数量低于真核生物,但其普遍性远超预期(图1)。在全局水平上,冗余基因数量与基因组大小呈显著正相关。更为关键的是,病原菌的冗余比例明显高于非病原菌,表明遗传信息的冗余很可能是病原菌重要的适应进化策略。
通过对冗余基因功能的系统注释,研究团队发现冗余基因并非随机扩增,而是与生态位适应密切相关。例如,海洋细菌富含运动相关冗余基因以适应流体环境;陆地细菌则更依赖信号转导和应激响应相关冗余;植物根际等相关微生物往往拥有更多冷休克蛋白的冗余以应对温度波动;鸟类宿主微生物则强化脂肪酸代谢相关冗余来适应较高体温。这些结果表明,冗余基因为细菌提供了可塑性,使其能够迅速应对不同的生态压力。
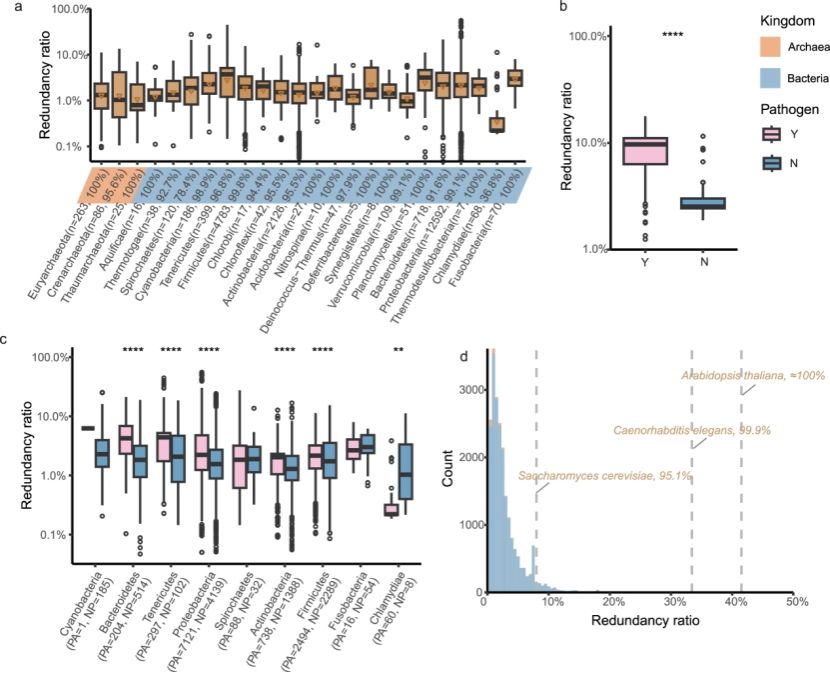
图1. 冗余基因在原核生物基因组中的普遍性
2. 冗余基因倾向保留功能,并与翻译起始信号的调控序列共同复制
在进化层面,研究团队观察到冗余基因比例随系统发育距离增加而上升,说明冗余在宏观进化尺度上不断积累(图2)。此外,冗余基因的多数副本具备完整的翻译起始信号,其表达调控结构也被同步复制,表明冗余基因不仅保留下来,而且大多数保持功能性。不同于真核生物中冗余基因普遍较高的假基因化率,原核生物对冗余基因这一手段表现出更强的保留与利用倾向,体现其“高效且灵活”的进化策略。
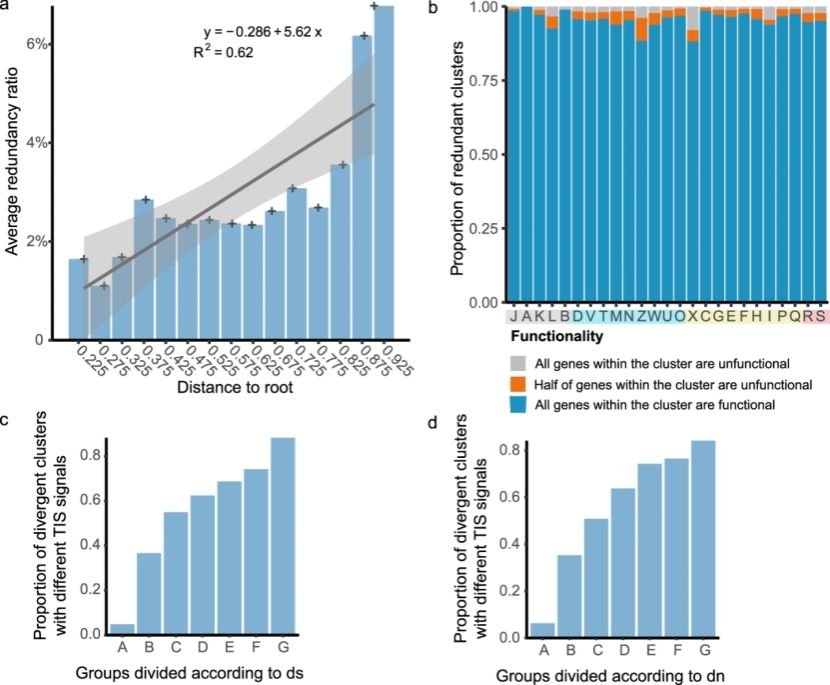
图2. 原核生物基因组中冗余基因的进化策略
3. 临床案例:冗余基因如何增强病原致病性
研究团队分析了898株Enterobacter cloacae complex(阴沟肠杆菌复合群,ECC),发现其冗余基因同样与病原相关功能高度相关。尤其是E. hormaechei,其冗余基因显著多于同属其它物种,许多冗余基因与重金属耐受(如铜、银、汞)、毒素-抗毒素系统和厌氧代谢相关。这些冗余基因大量来源于质粒与水平基因转移,反映其在医院环境中通过增强抗性与竞争力而占据生态优势。
为了更具体地评估冗余基因在临床感染中的作用,朱怀球团队与合作者肖永红团队对ICU(重症监护室)病房在6个月内采集的69株Acinetobacter baumannii(鲍曼不动杆菌)进行了全基因组测序及时间序列分析。这些菌株来自患者的呼吸道、尿路、血液、粪便以及病房环境表面。研究发现,与其他来源相比,尿路感染菌株中有两个关键的乙醇脱氢酶家族基因——frmA与ZADH的双拷贝冗余显著集中。进一步分析显示,这些冗余基因通过基因岛和插入序列(IS元件)引入,属于典型的水平基因转移事件。在两位ICU患者的纵向样本中,研究团队观察到A. baumannii常先在呼吸道定植,随后获得frmA与ZADH冗余基因后扩散至尿路并建立持久感染。更重要的是,冗余基因及其翻译起始信号在体内感染过程中保持稳定,说明双拷贝frmA基因可能提供了明确的选择优势。
为了验证这一点,研究团队开展了小鼠腹腔感染实验。结果显示,携带双拷贝frmA的菌株表现出显著更强的致死性,所有感染小鼠在24小时内死亡;而单拷贝菌株则表现出更低的致病性。同时,双拷贝菌株产生的生物膜量显著增加。进一步的质粒克隆与表达实验再次验证,提升frmA副本数或表达水平均能增强生物膜形成。这直接证明了冗余基因能够通过增加剂量效应提升病原菌毒力与定植能力。
总的来说,本研究首次大规模构建了原核生物冗余基因图谱,并通过临床样本与动物实验链式验证了冗余基因在病原菌适应性进化与临床感染中的关键作用。冗余基因不仅是细菌快速进化的重要遗传来源,还可能是未来病原控制与抗菌策略的新靶点。
北京大学未来技术学院、定量生物学中心朱怀球教授课题组已毕业博士王霈虹、已毕业博士郭倩、已出站博士后江小青、浙江大学医学院附属第一医院博士生路平为本文的共同第一作者。北京大学朱怀球教授、浙江大学肖永红教授为本文的共同通讯作者。本工作得到了国家自然科学基金委(No. 32070667, 82202588, 32300078)、科技部国家重点研发计划(No. 2017YFC1200205, 2021YFC2300300)的经费资助与北京大学国家生物医学成像中心生物医学计算平台、北京大学高性能计算平台的技术支持。
本文链接:https://www.nature.com/articles/s41467-025-65840-7
Science Magazine评述链接:https://scienmag.com/gene-redundancy-unlocks-pathogen-evolution-and-infection/







